Since the transition to genomics, data has gotten larger and larger.
Growth of Genbank
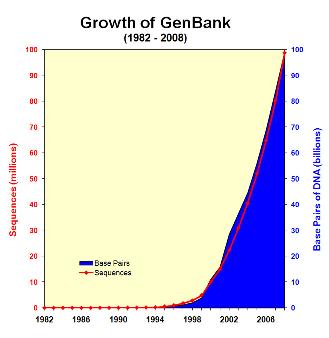
Fortunately, until the mid-90’s, disk size, network bandwidth & CPU speeds were increasing much faster so everyone was pretty happy. Now with HTS, some bottlenecks are starting to crop up.
Moore’s Law predicts that CPU power increases by 60% per year, but CPU clock speeds are essentially static and computational power is now being increased by multicore CPUs and GPUs which require considerably different programming techniques.
Network speeds are fairly fast, but are not increasing very fast locally because copper is reaching its upper limit and even if it increases a few more steps it will be at very short distances. Optical networks hold huge promise, but the technology is comparatively expensive and tempermental.
Disk capacity is continuing to increase exponentially as well, but large RAIDs are starting to approach the Uncorrectable Bit Error Rate problem and so the amount of data that we can store in contiguous devices is starting to hit a limit. And if you want that data backed up, the problem/expense doubles.
So what do we do? Learn to live with it.
It really boils down to the question above.
-
What is the minimum data you need to keep around?
-
Do you really need to back up everything?
-
Where do you put this data?
-
on large RAIDs, preferably RAID6 (any OS) or ZFS (Solaris only) or be prepared to give a LOT of money to BlueArc, Isilon, NetApp, EMC, Sun, IBM, etc.
-
Open Source Parallel File systems are starting to be reliable enough to use in production as well (Lustre, PVFS, Gluster, etc)
-
if on your Desktop, use reliable disks & RAID them if possible.
-
How do you move this data?
Note that for most HTS data, using encryption is not ncessary (do you care if someone intercepts your short-read file?) so for most files, don’t bother with encryption. On the other hand, compression can help a lot on slower connections (plaintext sequence data is compressible to 1/4 to 1/3 the uncompressed size.
A somewhat wooden analogy

The above image of a spinning wooden dish on a lathe is a (strained) analogy to a disk drive. The faster the dish spins, the more wood (data) can be scraped off it per second; the closer to the outside, the more wood (data) can be scraped off per second. If you have multiple lathes (disks), each additional lathe increases the amount of wood (data) that can be scraped off per second.
There is hope, especially for some types of data.
3.1. Types of data
3.1.1. Plaintext
-
ASCII plaintext vs binary vs MS Word doc format For editing plaintext files, use a plaintext editor, such as Notepad for Windows or Jedit (excellent, for all platforms).
-
Mac and Linux plaintext now use the same end-of-line character (line feed; \n); Windows is still different (carriage return + line feed; \r\n)
3.1.2. Structured Text
3.1.3. Binary
-
images
-
compressed data
-
executables, object files, libraries
-
specially constructed application-specific data.
3.1.4. Databases
-
flatfile (FASTA sequences)
-
relational (MySQL, PostgreSQL)
-
key:value type DBs (color:red, fruit:bananas)
3.1.5. Self-describing Data
Structured (strided, self-describing) binary files (such as NetCDF, HDF5, & BioHDF).
Self-describing Data Formats are a special case of data file and one that holds great promise for large Biological data. It has been used for many kinds of large data for more than a decade in the Physical Sciences and since part of Biology is now transitioning to a Physical Science, it becomes appropriate to use their proven approaches.
-
can represent N-dimensional data
-
have unlimited extents (such as open-ended time series)
-
are internally like an entire filesystem, so groups of related data can be bundled together.
-
are indexed (or strided) so that offsets to a particular piece of data can be found very quickly (microseconds).
-
are dynamically ranged so the file uses only as many bits to represent a range of data as it really needs. ('int’s can be represented as bits, depending on the data range)
The BioHDF project is already starting to use this approach, specifically for representing HTS data and we should start seeing code support later this year. It happens that Charlie Zender (author of nco, a popular utility suite for manipulating NetCDF files) is in UCI’s ESS dept.
There is another technology that has been used quite successfully in the Climate Modelling field which is the OpenDAP Server. This software keeps the data on a central server and allows researchers to query it to return various chunks of data and to transform them in various ways. In some ways it’s very much like a web server such as the UCSC Genome Center’s Browser system, but it allows much more computationally heavy transforms.
4.1. Basic utilities
The following are basic Linux utilities.
4.1.1. ls
ls lists the files in a directory
4.1.2. grep
4.1.3. head & tail
head shows you the top of a file. tail shows you the bottom of the file.
4.1.4. cut, scut & cols
cut and scut let you slice out and re-arrange columns in a file, spearated by a defined delimiter.
cols lines up those files so you can make sense of them.
4.2. Applications on the BDUC Cluster
*R/2.10.0 *gaussian/3.0 *mosaik/1.0.1388 readline/5.2
*R/2.11.0 gnuplot/4.2.4 mpich/1.2.7 *samtools/0.1.7
*R/2.8.0 *gromacs_d/4.0.7 mpich2/1.1.1p1 scilab/5.1.1
*R/dev *gromacs_s/4.0.7 *msort/20081208 sge/6.2
*abyss/1.1.2 hadoop/0.20.1 *namd/2.6 *soap/2.20
antlr/2.7.6 hdf5-devel/1.8.0 *namd/2.7b1 sparsehash/1.6
*autodock/4.2.3 hdf5/1.8.0 ncl/5.1.1 sqlite/3.6.22
*bedtools/2.6.1 imagej/1.41 nco/3.9.6 subversion/1.6.9
*bfast/0.6.3c interviews/17 netCDF/3.6.3 *sva/1.02
boost/1.410 java/1.6 neuron/7.0 *tacg/4.5.1
*bowtie/0.12.3 *maq/0.7.1 *nmica/0.8.0 tcl/8.5.5
*bwa/0.5.7 matlab/R2008b octave/3.2.0 tk/8.5.5
*cufflinks/0.8.1 matlab/R2009b open64/4.2.3 *tophat/1.0.13
fsl/4.1 *mgltools/1.5.4 openmpi/1.4 *triton/4.0.0
*gapcloser *modeller/9v7 python/2.6.1 visit/1.11.2
* of possible interest to Biologists.
example:
$ module load abyss
ABySS - assemble short reads into contigs
See: /apps/abyss/1.1.2/share/doc/abyss/README
or 'ABYSS --help'
or 'man /apps/abyss/1.1.2/share/man/man1/ABYSS.1'
or 'man /apps/abyss/1.1.2/share/man/man1/abyss-pe.1'
Also, ClustalW2/X, SATE (including MAFFT, MUSCLE, OPAL, PRANK, RAxML), readseq, phylip, etc.
Some other Genomic Visualization tools are listed here.
(Some of them are installed on BDUC; some are web tools; some still need to be installed.
Sometimes what you need to do just isn’t handled by an existing utility and you need to write it yourself. For those situations which call on data mangling/massaging, I’d recommend either Perl & BioPerl (or Python & BioPython).
Perl has an elegant syntax for handling much of the simple line-based data mangling you might run into:
#!/usr/bin/perl -w
while (<>) { # while there's still more lines to process
chomp; # chew off the 'newline' character
$N = @L = split; # split all the columns and place them into the array named 'L'
<your code> # do what you need to do to the data
} # continue to the end of the file.
For genomic data analysis, I’d very strongly recommend R & BioConductor. I wrote a very gentle introduction to R and cite many more advanced tutorials therein. Both R and BioConductor have been advancing extremely rapidly recently and are now the de facto language of Genomics as well as Statistics. While it lacks a GUI, R has many advanced visualization tools and graphic output.
Also, there is a website called Scriptome which has started to gather and organize scripts for the slicing, dicing, reformatting, and joining of sequence and other data related to Bioinfo, CompBio, etc.
6.1. BDUC Advancement
6.2. Latest Version
The latest version of this document moo:~/public_html/Bio/BigBioData.html[should be here].
